



Über hTTP
Upshaw Schulman Syndrome
Upshaw-Schulman Syndrome
Klassifikation und externe Ressourcen
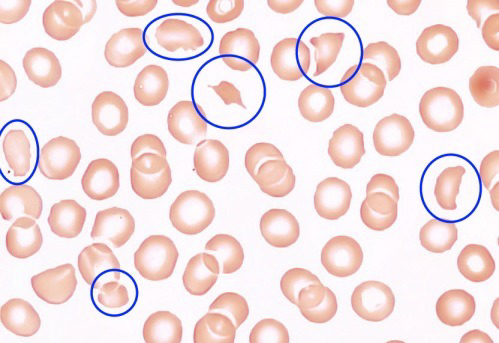
Blutausstrich eines TTP Patienten unter dem Mikroskop mit Schistozyten (Fragmentozyten) blau markiert.
| ICD-10 | M31.1 (ILDS M31.110) |
| ICD-9 | 446.6 |
| OMIM | 274150 |
| DiseasesDB | 13052 |
| MedlinePlus | 000552 |
| eMedicine | emerg/579 neuro/499 med/2265 |
| MeSH | D011697 |
Wikipedia Artikel (Stand April 2017). Original geschrieben vom TTP Forschungs Team in Bern, Schweiz
Das Upshaw-Schulman-Syndrom (USS) ist eine seltene Blutgerinnungskrankheit und entspricht der vererbten Form der thrombotisch thrombozytopenischen Purpura (TTP). Patienten mit USS haben zu wenig ADAMTS13-Protease, dadurch bleiben die Multimere des ultralangen Von-Willebrand-Faktors (ULVWF) im Blut bestehen, welche eine thrombotische Mikroangiopathie mit Gefäßverschlüssen in den kleinen Blutgefäßen auslösen [1][2]. Diese Gefäßverschlüsse verhindern eine genügende Durchblutung des dahinter liegenden Gewebes, welches infolgedessen geschädigt wird. Die Symptome bei akutem USS sind sehr variabel. In den meisten Fällen besteht eine thrombozytopenische mikroangiopathische hämolytische Anämie (MAHA) mit Schistozyten im Blutausstrich [3], Fieber und ischämisch bedingten Organschäden in Hirn, Niere und Herz.
Epidemiologie
Die Häufigkeit einer TTP beträgt 1,7 bis 4,5 Million pro Jahr [4][5]. Die meisten TTP-Fälle sind der autoimmunen TTP zuzuschreiben und werden durch Autoantikörper verursacht, welche die ADAMTS13-Protease blockieren [1][6]. Nur etwa 5 % der TTP-Fälle werden durch das USS verursacht [7]. Die genaue Prävalenz des USS konnte wegen seiner Seltenheit noch nicht berechnet werden.
Das Upshaw Schulman Syndrom wird autosomal rezessiv vererbt. Es wird häufiger durch compound heterozygote Mutationen als durch homozygote Mutationen verursacht [7]. Das Manifestationsalter ist variabel und reicht vom Neugeborenen- bis ins hohe Erwachsenenalter [1]. Die Wahrscheinlichkeit eines wiederkehrenden Verlaufs ist sehr individuell [1][6]. Die Schwere der Krankheit kann mit früh gestellter Diagnose und gegebenenfalls prophylaktischer Therapie gemindert werden [2].
Aetiologie
Genetische Mutationen
Das ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif 13) Gen ist auf dem Chromosom 9q34 codiert und enthält 29 Exone [8]. Die ADAMTS13-Protease besteht aus 1427 Aminosäuren die folgende Domänen bilden [1][8]:
- Der „signal peptide region“ wird Einfluss auf die Sekretion, Faltung und Stabilität der ADAMTS13-Protease zugeschrieben [9]. Sie wirkt an Phospholipiden der Zellmembran und Proteinen der Sekretionskomplexen in den ADAMTS13 produzierenden Zellen [9].
- Die „metalloprotease-domain“ ist der aktive Teil des ADAMTS13, die den VWF an dessen A2-Domäne zwischen den Aminosäuren Tyrosin1605-Methionin1606 spaltet [10].
- Die „disintegrin-domain“ zusammen mit dem „Thrombospondin-1 repeat“ (TSP-1-repeat) und der anschließenden „Cystein-rich domain“ und „Spacer-domain“ werden für die Substraterkennung und Spaltung der VWF [9] A2 Domäne [10] benötigt. Als erstes erkennt die „Spacer-Domain“ den VWF und erhöht die Affinität von ADAMTS13 zu VWF. Danach reagiert die „Disintegrin-like-domain“ mit einer niederaffinen Bindung. Zuletzt bindet die „Metalloprotease-domain“ an den VWF, wie ein dreiteiliger Reißverschluss [6].
- Die „TSP-1-repeats“ beeinflussen Interaktionen zwischen Proteinen und der extrazellulären Matrix [11].
- Die „Cystein-rich domain“ ist wichtig für das Andocken an Zellen, zum Beispiel an Integrine von bestimmten Zellmembranen [11].
- Die „CUB-domains“ gehen Verbindungen zwischen Proteinen des VWFs ein [11], die bei hohen Scherkräften zugänglich sind [10]. Sie sind sowohl für das Binden als auch für das Spalten von VWF verantwortlich [9]. Zusätzlich sind sie an der ADAMTS13-Sekretion aus den Zellen beteiligt [12].
Mutationen, die USS verursachen kommen in allen Domänen vor [10]. Bei USS wird vor allem die Sekretion von ADAMTS13 gestört, wobei die ADAMTS13-Aktivität zusätzlich herabgesetzt oder verloren gehen kann [9]. Momentan sind über 120 USS verursachende Mutationen und zahlreiche Single nucleotide polymorphismen (SNP) bekannt [13][14]. Die verschiedenen SNPs können, je nach Kombinationen, die ADAMTS13-Protease-Aktivität verstärken oder abschwächen [9][8]. Patienten mit einer Restaktivität der ADAMTS13-Protease neigen zu einem späteren Krankheitsbeginn [13].
Die Funktion von ADAMTS13 und Pathogenes von USS
Die Familie der ADAMTS-Proteasen beinhaltet Enzyme zur Kollagenprozessierung, Spalten von interzellulärer Matrix, Hemmung der Angiogenese und zur Antikoagulation. Die ADAMTS13 ist eine Zink-Metalloprotease. Sie wird hauptsächlich in Leberzellen und Endothelzellen hergestellt, wurde aber auch in anderen Zellen, wie Thrombozyten, Nierenzellen und Hirnzellen gefunden [10][15][16]. Die einzige bekannte Funktion von ADAMTS13 ist das Spalten der VWF-Multimere [17]. Die messbare Plasmahalbwertszeit im Blut von ADAMTS13 beträgt ca. 2 bis 4 Tage, wobei die protektive Wirkung länger anzuhalten scheint [18][19].
Ein Patient mit USS hat meist eine schwere ADAMTS13-Aktivitätsverminderung von weniger als 10 % der normalen Aktivität [6]. Je nach vorliegenden Mutationen kann in diesem Bereich eine Restfunktion erhalten bleiben [9][8][13].
Eine tiefe ADAMTS13-Aktivität reicht oft nicht aus um eine (erste) TTP-Episode auszulösen. Ein akuter TTP-Schub bei USS-Patienten wird häufig durch einen umweltbedingten Auslöser initiiert [1][10][8]. Bekannte Auslöser sind Infektionen (einschließlich leichter Infektionen der oberen Luftwegen), Schwangerschaft [20], erhöhter Alkoholkonsum[18] und Medikamente [1][6]. In diesen Situationen wird VWF aus den Speicherorganellen (zum Beispiel den Palade-Weibel-Körperchen, Plättchengranula) freigesetzt. Dadurch steigt der VWF-Gehalt des Blutes an und mehr ADAMTS13 wird benötigt um eine spontane Gerinnung des Blutes in den kleinen Blutgefäßen zu verhindern. Der Mangel an ADAMTS13-Aktivität in USS-Patienten führt in Folge zu einer TTP-Episode [3][11].
Pathologie
Nach seiner Freisetzung ist ADAMTS13 entweder an die Gefäßwand (Endothel) gebunden oder frei im Plasma [3]. Die starken Scherkräfte in kleinen Blutgefäßen ziehen den zusammengezogenen kugeligen VWF in seine lineare Form [10]. Die aktive Bindungsdomäne des linearen VWF liegen in dieser Form frei an der Oberfläche, und können die Blutgerinnung auslösen [10]. Diese Bindungsdomänen verbinden Plättchen mit Läsionen an Blutgefäßen und vernetzen VWF untereinander, damit ein Blutgerinnsel gebildet werden kann [10]. Die ULVWF-Multimere haben eine erhöhte Reagibilität und Bindungskapazitäten, was zu spontanen Gefäßverschlüssen führt. Ist genügend ADAMTS13 vorhanden, kann dieser den linearen ULVWF in seine normale Größe spalten [3]. Normal großer VWF ist weniger bindungsfreudig und nur notwendige Blutgerinnsel werden gebildet [3].
Symptome
Das klinische Bild einer TTP ist sehr variabel. Der Patient/ die Patientin sucht häufig den Arzt/ die Ärztin auf mit Symptomen die als Folge der häufig tiefen Plättchenzahl auftreten, wie eine Purpura (ca. in 90 % der Patienten), Ekchymosen und Hämatomen [21]. Daneben können noch weitere Symptome, die als Folge einer mikroangiopathischen hämolytischenAnämie vorhanden sind, auftreten, wie dunkler Urin, (milder) Ikterus, Müdigkeit und Blässe. Viele Patienten/Patientinnen haben zusätzlich verschieden ausgeprägte neurologische Symptome, wie Kopfschmerzen, Bewegungsstörungen, Sprachstörungen, Schlaganfälle und Bewussteinstrübungen bis hin zu komatösen Zuständen. Die Symptome können zusätzlich während eines Schubes vorübergehend verschwinden und wieder erscheinen. Weitere unspezifische Befunde sind Unwohlsein und Gelenk- oder Muskelschmerzen. Schwere Beeinträchtigungen von Herz oder Lunge sind selten, wobei häufig Veränderungen in den Organsystemen messbar sind (z. B. EKG-Abnormitäten) [11].
Diagnose
Die Diagnose TTP wird anhand der klinischen Zeichen und Symptome bei gleichzeitigem Vorliegen einer Thrombozytopenie (Thrombozytenzahl kleiner 100 × 109/l, häufig kleiner 20 × 109/l), mikroangiopathischen hämolytischen Anämie mit Schistozyten im Blutausstrich, einem negativen direkten Antiglobulin Test (Coombs-Test) und erhöhten Hämolysemarkern (z. B. totales Bilirubin, LDH, freies Hämoglobin, tiefes Haptoglobin) nach Ausschluss anderer möglichen Ursachen gestellt [1][2].
Folgende Diagnosen, die USS gleichen, werden üblicherweise ausgeschlossen [1][2]: Fulminante Infektion, disseminierte intravasale Koagulopathie (DIC), autoimmunhämolytische Anämie, Evans-Syndrom, autoimmune atypische oder postinfektiöse Form des hämolytischen urämischen Syndroms (HUS), HELLP-Syndrom (hemolysis, elevated liver enzymes, low platelets – Syndrom), Präeklampsie und Eklampsie, Heparin-induzierte Thrombozytopenie (HIT), (metastasierter) Krebs, Nierenschäden, Antiphospholipid-Antikörper-Syndrom, und Nebenwirkungen einer Knochenmarkstransplantation [1][2].
Die schwangerschaftsassoziierten Krankheitsbilder, wie Präeklampsie, Eklampsie sowie das HELLP-Syndrom können mit dem einer TTP-Episode überlappen, da sie selbst einen TTP-Schub auslösen können [22] und bedürfen besonderer Aufmerksamkeit.
Patienten mit fulminanten Infektionen, disseminierter intravasaler Gerinnung (DIC), HELLP-Syndrom, Pankreatitis, Leberkrankheiten oder anderen Entzündungen können eine gesenkte ADAMTS13-Aktivität aufweisen. Unter diesen Umständen lösen sie sehr selten eine schwere krankheitsrelevante Verminderung der ADAMTS13-Aktivität unter 10 % aus [2][23].
Ein schwerer ADAMTS13-Mangel <5 % oder <10 % (je nach Definition[1][24]) ist der Beweis für eine TTP. [25][18] Die ADAMTS13-Tests messen dabei direkt oder indirekt die VWF Spaltprodukte. Die ADAMTS13-Aktivität sollte aus einer Blutentnahme vor Therapiebeginn gemessen werden, um eine falschhohe ADAMTS13-Aktivität auszuschließen [2].
Bei schwerem ADAMTS13-Mangel muss die autoimmune Form von der angeborenen Form der TTP, dem USS, unterschieden werden [1]. Die Antikörper werden entweder direkt mit einem sogenannten ELISA-Test oder indirekt, durch die Testung auf einen funktionalen Inhibitor gesucht. Bei autoimmuner TTP kann die Menge der Antikörper im Blut schwanken, daher werden bei negativem Testresultat eine zweite Messungen zur Bestätigung während einer TTP-Episoden-freien Phase durchgeführt [2]. Die Bestätigung eines schweren ADAMTS13-Mangels in Abwesenheit von Antikörpern stellt meist die Indikation für eine Genanalyse zum Beweisen einer USS-verursachenden Mutation dar.
In unklaren Fällen kann ein sogenanntes Plasma-Infusions-Trial durchgeführt werden. Hierbei kann die dosisabhängige ADAMTS13-Aktivität in der für USS typischen Halbwertszeit von 2 bis 4 Tagen bestätigt werden. Partieller oder schwerer ADAMTS13-Mangel bei einem Verwandten ersten Grades ist ebenfalls ein starker Hinweis auf USS [1][2][6].
Therapie
Eine TTP-Episode benötigt eine sofortige Behandlung [1][2]. Die Standardtherapie besteht aus täglichem Ersatz der ADAMTS13-Protease durch Plasmainfusion oder in schweren Fällen aus einer Plasmaaustausch-Therapie (PEX). Bei einer PEX wird das patienteneigene Plasma annähernd vollständig durch Spenderplasma ersetzt [26]. In beiden Fällen wird am häufigsten plättchenarmes FFP (Fresh-Frozen-Plasma) verwendet, wobei auch andere Plasmaprodukte, die ADAMTS13 enthalten eingesetzt werden können [3]. Der Vorteil einer PEX-Therapie gegenüber Plasmainfusionen wird dem Eliminieren der überschüssigen ULVWF-Multimere zugeschrieben [1]. Einige, meist milde Nebenwirkungen, wurden dabei beobachtet [2][26]. Die benötigte Anzahl Infusionen oder PEX zur Genesung variieren, wobei die Therapie des USS meistens weniger als eine Woche dauert[1][6]. Die Plasmatherapie kann beendet werden, wenn sich die Thrombozytenzahl normalisiert und über mehrere Tage stabil ist [1][2].
Vorbeugende Therapie
Nicht alle von USS betroffenen Patienten/Patientinnen benötigen eine regelmäßige vorbeugende Plasmainfusion. Diejenigen mit häufigen TTP-Schüben sind jedoch auf eine solche Behandlung angewiesen [27]. Eine Therapie mit Plasmainfusionen alle zwei bis drei Wochen kann solchen Schüben vorbeugen, wobei die Therapie jeweils individuell angepasst werden kann [18]. Milde Krankheitsverläufe ohne häufiges Wiederauftreten von TTP-Schüben bedürfen nur in speziellen Risikosituationen (wie oben erwähnt) Plasmainfusionen [27].
Ausblick
In den letzten Jahren konnten neue Entwicklungen in der TTP-Forschung beobachtet werden. Eine rekombinante ADAMTS13-Protease durchlief erste erfolgreiche Tests in Mäusen [28] und erste Tests an USS-Patienten/ Patientinnen wurden angekündigt. Daneben wurde ein weiteres Medikament vorgestellt, das die (UL)VWF-Interaktionen mit Blutplättchen reduziert und dadurch die überschießende Gerinnung bei einer TTP-Episode hemmt [29]. Neben verschiedenen multinationalen Datenbanken wurde ein weltweites Projekt zur Forschung an USS gestartet, welches Informationen über Patienten und deren Familienangehörige sammelt, um daraus neue Erkenntnisse zu Diagnose und Therapieoptimierung abzuleiten [30].
Geschichte

Die TTP als solche wurde zum ersten Mal 1947 beschrieben und nach ihrer Pathophysiologie benannt [18]. 1960 beschrieb Schulman den ersten USS-Patienten [1]. Upshaw berichtete 1978 von einer rezidivierenden TTP, die er während 11 Jahren begleitet hatte [31]. Upshaw beschrieb dabei die Parallelen der beiden Fälle und vermutete als Ursache ebenfalls das Fehlen eines Plasmafaktors [1][31]. Ein Jahr später wurde das Syndrom erstmals Upshaw-Schulman-Syndrom genannt [32]. 1996 wurde das Fehlen des VWF-spaltenden Enzyms als Ursache von USS erkannt [17][33][34]. 2001 hat man diese Protease ADAMTS13 benannt, auf dem Chromosom 9q34 lokalisiert und erste krankheitsverursachende Mutationen bestimmt [1][19].
Referenzen
- J. Evan Sadler: Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. In: Blood. Band 112, Nr. 1, 1. Juli 2008, S. 11–18, doi:10.1182/blood-2008-02-078170, PMID 18574040.
- Ravi Sarode, Nick Bandarenko, Mark E. Brecher, Joseph E. Kiss, Marisa B. Marques, Zbigniew M. Szczepiorkowski, Jeffrey L. Winters: Thrombotic thrombocytopenic purpura: 2012 American Society for Apheresis (ASFA) consensus conference on classification, diagnosis, management, and future research. In: Journal of Clinical Apheresis. Band 29, Nr. 3, 1. Juni 2014, S. 148–167, doi:10.1002/jca.21302.
- Joel L. Moake: Thrombotic Microangiopathies. In: New England Journal of Medicine. Band 347, Nr. 8, 22. August 2002, S. 589–600, doi:10.1056/NEJMra020528, PMID 12192020.
- D. R. Terrell, L. A. Williams, S. K. Vesely, B. Lämmle, J. a. K. Hovinga, J. N. George: The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. In: Journal of Thrombosis and Haemostasis. Band 3, Nr. 7, 1. Juli 2005, S. 1432–1436, doi:10.1111/j.1538-7836.2005.01436.x.
- Jessica A. Reese, Darrshini S. Muthurajah, Johanna A. Kremer Hovinga, Sara K. Vesely, Deirdra R. Terrell, James N. George: Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. In: Pediatric Blood & Cancer. Band 60, Nr. 10, Oktober 2013, S. 1676–1682, doi:10.1002/pbc.24612, PMID 23729372.
- J. T. B. Crawley, M. A. Scully: Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies. In: Hematology. Band 2013, Nr. 1, 1. Dezember 2013, S. 292–299, doi:10.1182/asheducation-2013.1.292.
- Reinhard Schneppenheim u. a.: A common origin of the 4143insA ADAMTS13 mutation. In: Thrombosis and Haemostasis. Band 96, Nr. 1, 14. Juni 2006, S. 3–6, doi:10.1160/TH05-12-0817.
- R. S. Camilleri, H. Cohen, I. J. Mackie, M. Scully, R. D. Starke, J. T. B. Crawley, D. A. Lane, S. J. Machin: Prevalence of the ADAMTS-13 missense mutation R1060W in late onset adult thrombotic thrombocytopenic purpura. In: Journal of Thrombosis and Haemostasis. Band 6, Nr. 2, 1. Februar 2008, S. 331–338, doi:10.1111/j.1538-7836.2008.02846.x.
- Barbara Plaimauer u. a.: Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. In: Blood. Band 107, Nr. 1, 1. Januar 2006, S. 118–125, doi:10.1182/blood-2005-06-2482, PMID 16160007.
- Stefano Lancellotti, Maria Basso, Raimondo De Cristofaro: Proteolytic Processing of Von Willebrand Factor by Adamts13 and Leukocyte Proteases. In: Mediterranean Journal of Hematology and Infectious Diseases. Band 5, Nr. 1, 2. September 2013, ISSN 2035-3006, doi:10.4084/MJHID.2013.058, PMID 24106608.
- Miriam Biol, Marina Noris, Giuseppe Remuzzi: Thrombotic Thrombocytopenic Purpura-Then and Now. In: Seminars in Thrombosis and Hemostasis. Band 32, Nr. 2, März 2006, S. 81–89, doi:10.1055/s-2006-939763.
- Zhou Zhou, Hui-Chun Yeh, Hua Jing, Christina Wang, Zhenyin Tao, Huiwan Choi, Khatira Aboulfatova, Renhai Li, Jing-Fei Dong: Cysteine residues in CUB-1 domain are critical for ADAMTS13 secretion and stability. In: Thrombosis and Haemostasis. Band 105, Nr. 1, Januar 2011, S. 21–30, doi:10.1160/TH10-07-0446, PMID 20886194.
- Luca A. Lotta u. a.: Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura. In: Blood. Band 120, Nr. 2, 12. Juli 2012, S. 440–448, doi:10.1182/blood-2012-01-403113, PMID 22529288.
- Luca A. Lotta, Isabella Garagiola, Roberta Palla, Andrea Cairo, Flora Peyvandi: ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. In: Human Mutation. Band 31, Nr. 1, Januar 2010, S. 11–19, doi:10.1002/humu.21143, PMID 19847791.
- Minola Manea, AnnCharlotte Kristoffersson, Reinhard Schneppenheim, Moin A. Saleem, Peter W. Mathieson, Matthias Mörgelin, Peter Björk, Lars Holmberg, Diana Karpman: Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura. In: British Journal of Haematology. Band 138, Nr. 5, 1. September 2007, S. 651–662, doi:10.1111/j.1365-2141.2007.06694.x.
- Ryoji Tauchi, Shiro Imagama, Tomohiro Ohgomori, Takamitsu Natori, Ryuichi Shinjo, Naoki Ishiguro, Kenji Kadomatsu: ADAMTS-13 is produced by glial cells and upregulated after spinal cord injury. In: Neuroscience Letters. Band 517, Nr. 1, 23. Mai 2012, S. 1–6, doi:10.1016/j.neulet.2012.03.002.
- M. Furlan, R. Robles, B. Lamie: Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. In: Blood. Band 87, Nr. 10, 15. Mai 1996, S. 4223–4234, PMID 8639781.
- Johanna A. Kremer Hovinga, Bernhard Lämmle: Role of ADAMTS13 in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. In: ASH Education Program Book. Band 2012, Nr. 1, 12. August 2012, S. 610–616, PMID 23233642.
- Miha Furlan, Rodolfo Robles, Beat Morselli, Pierre Sandoz, Bernhard Lämmle: Recovery and Half-Life of von Willebrand Factor-Cleaving Protease after Plasma Therapy in Patients with Thrombotic Thrombocytopenic Purpura. In: Thromb Haemost. Band 81, Nr. 1, 1999, S. 8–13.
- Y. Jiang u. a.: Pregnancy outcomes following recovery from acquired thrombotic thrombocytopenic purpura. In: Blood. Band 123, Nr. 11, 13. März 2014, S. 1674–1680, doi:10.1182/blood-2013-11-538900.
- James N. George, Qiaofang Chen, Cassie C. Deford, Zayd Al-Nouri: Ten patient stories illustrating the extraordinarily diverse clinical features of patients with thrombotic thrombocytopenic purpura and severe ADAMTS13 deficiency. In: Journal of Clinical Apheresis. Band 27, Nr. 6, 1. Januar 2012, S. 302–311, doi:10.1002/jca.21248.
- Eriko Yamashita, Hidetaka Okada, Hirokazu Yorioka, Shinya Fujita, Kenichiro Nishi, Yutaka Komiyama, Hideharu Kanzaki: Successful management of pregnancy-associated thrombotic thrombocytopenic purpura by monotoring ADAMTS13 activity. In: Journal of The Journal of Obstetrics and Gynaecology Research. Band 38, Nr. 3, 1. März 2012, S. 567–569, doi:10.1111/j.1447-0756.2011.01742.x
- Masahito Uemura, Yoshihiro Fujimura, Saiho Ko, Masanori Matsumoto, Yoshiyuki Nakajima, Hiroshi Fukui: Determination of ADAMTS13 and Its Clinical Significance for ADAMTS13 Supplementation Therapy to Improve the Survival of Patients with Decompensated Liver Cirrhosis. In: International Journal of Hepatology. Band 2011, 18. Juli 2011, S. e759047, doi:10.4061/2011/759047.
- James N. George, Zayd L. Al-Nouri: Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. In: Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. Band 2012, 2012, S. 604–609, PMID 23233641.
- Hochspringen ↑A. Tripodi u. a.: Second international collaborative study evaluating performance characteristics of methods measuring the von Willebrand factor cleaving protease (ADAMTS-13). In: Journal of thrombosis and haemostasis: JTH. Band 6, Nr. 9, September 2008, S. 1534–1541, doi:10.1111/j.1538-7836.2008.03099.x, PMID 18662260.
- Gail A. Rock, Kenneth H. Shumak, Noel A. Buskard, Victor S. Blanchette, John G. Kelton, Rama C. Nair, Robert A. Spasoff: Comparison of Plasma Exchange with Plasma Infusion in the Treatment of Thrombotic Thrombocytopenic Purpura. In: New England Journal of Medicine. Band 325, Nr. 6, 8. August 1991, S. 393–397, doi:10.1056/NEJM199108083250604, PMID 2062330.
- Paul Knöbl: Inherited and acquired thrombotic thrombocytopenic purpura (TTP) in adults. In: Seminars in Thrombosis and Hemostasis. Band 40, Nr. 4, Juni 2014, S. 493–502, doi:10.1055/s-0034-1376883, PMID 24802084.
- Alexandra Schiviz, Kuno Wuersch, Christina Piskernik, Barbara Dietrich, Werner Hoellriegl, Hanspeter Rottensteiner, Friedrich Scheiflinger, Hans Peter Schwarz, Eva-Maria Muchitsch: A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. In: Blood. Band 119, Nr. 25, 21. Juni 2012, S. 6128–6135, doi:10.1182/blood-2011-09-380535, PMID 22529289.
- Josefin-Beate Holz: The TITAN trial – Assessing the efficacy and safety of an anti-von Willebrand factor Nanobody in patients with acquired thrombotic thrombocytopenic purpura. In: Transfusion and Apheresis Science. Band 46, Nr. 3, Juni 2012, S. 343–346, doi:10.1016/j.transci.2012.03.027.
- M. Mansouri Taleghani u. a.: Hereditary thrombotic thrombocytopenic purpura and the hereditary TTP registry:. In: Hämostaseologie. Band 33, Nr. 2, 2013, S. 138–143, doi:10.5482/HAMO-13-04-0026.
- Jefferson D. Upshaw: Congenital Deficiency of a Factor in Normal Plasma That Reverses Microangiopathic Hemolysis and Thrombocytopenia. In: New England Journal of Medicine. Band 298, Nr. 24, 15. Juni 1978, S. 1350–1352, doi:10.1056/NEJM197806152982407, PMID 651994.
- S. Rennard, S. Abe: Decreased cold-insoluble globulin in congenital thrombocytopenia (Upshaw-Schulman syndrome). In: The New England Journal of Medicine. Band 300, Nr. 7, 15. Februar 1979, S. 368, doi:10.1056/NEJM197902153000718, PMID 759902.
- H. M. Tsai: Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. In: Blood. Band 87, Nr. 10, 15. Mai 1996, S. 4235–4244, PMID 8639782.
- M. Furlan, R. Robles, M. Solenthaler, M. Wassmer, P. Sandoz, B. Lämmle: Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. In: Blood. Band 89, Nr. 9, 1. Mai 1997, S. 3097–3103, PMID 9129011.